据统计,人类蛋白中有接近80%蛋白无显著活性位点,传统上被认为是不可成药靶点。传统的靶标大部分是具有明确活性位点的蛋白,且适合结合小分子,并主要通过占据活性位点的药理学作用模式 (MOA) 来控制蛋白功能。这种方法虽然简单,但是并不能应用于所有生物靶标,包括那些缺乏酶活、曾被认为是「不可成药」的靶标,以及通过突变反复耐药、现阶段无药可治的靶标。因此长期以来,靶向蛋白的药物研发受限于有限的蛋白种类,极大地限制了药物发展。
自耶鲁大学的Craig Crews教授团队首次报道了小分子PROTAC后的十年间,PROTAC领域飞速发展。药物靶标的格局也因此发生了重大变化,从传统药物靶标转向更具挑战性的靶标。
纵观PROTAC技术发展历史,2001年首个PROTAC分子PROTAC-1诞生,是由PROTAC技术先驱Crews及其同事报道;2008年,第一个小分子PROTAC诞生,同样由Crews课题组设计合成,是由靶向MDM2 E3泛素连接酶的小分子抑制剂nutlin和AR小分子配体以及中间的PEG Linker构成;之后,研究者开发出以CRL4CRBN、CRL2VHL、cIAPE3泛素连接酶配体的各种小分子PROTACs;再之后,Arvinas公司研发的ARV-110和ARV-471两个PROTAC分子进入临床实验,分别用于治疗前列腺癌和乳腺癌。
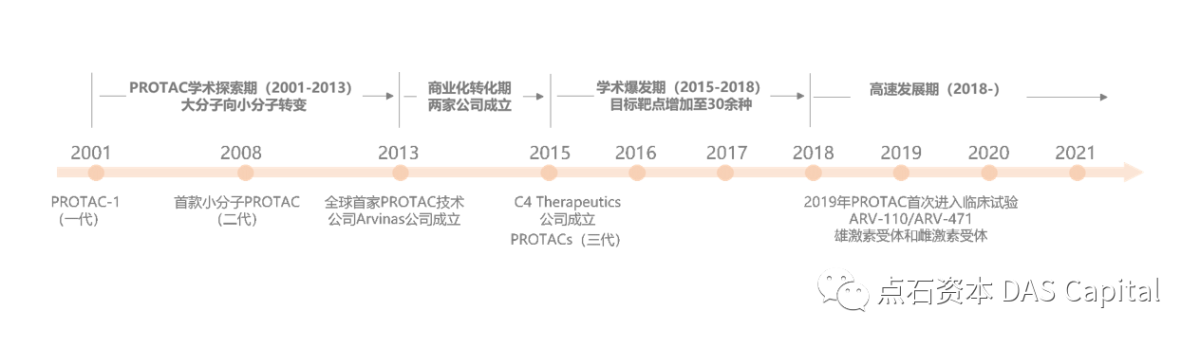
图:PROTAC里程碑时间点
PROTAC 技术的出现,以其颠覆性的设计、颠覆性的作用机制挑战了「不可成药」靶点,使耐药患者获得新一代治疗药物。近两年来,PROTAC 技术进入高速发展阶段,为生物医药研发开拓了一片新的领域。
蛋白质是细胞内重要的生物大分子,参与多种生命活动,包括酶和激素的功能、运动、运输、免疫反应等。以往,人们非常关注蛋白质在细胞内是如何合成的,但对于蛋白质在细胞内是如何降解的却少有人关注。以色列科学家Aaron Ciechanover、Avram Hershko和美国科学家Irwin Rose发现了泛素介导的蛋白质降解机制,共同获得了2004年诺贝尔化学奖。
泛素本身也是一种多肽,由76个氨基酸残基组成,序列高度保守,且在多种真核生物中普遍存在。泛素的主要功能是标记需要分解的蛋白质,当泛素连接到蛋白上后,会导致这些蛋白被运送到蛋白酶体中进行降解,实现细胞内蛋白的平衡。总的来说,泛素参与了细胞周期、增殖、凋亡、分化、转移、基因表达、转录调节、信号传递、损伤修复、炎症免疫等几乎一切生命活动的调控。
泛素化,主要由3种酶催化,即E1泛素活化酶、E2泛素结合酶、E3连接酶。在ATP的作用下,泛素会被激活,先形成泛素-腺苷酸复合物,再被转移到泛素活化酶E1上;随后,E1将活化的泛素转移到泛素结合酶E2上;最终,泛素连接酶E3将泛素转移到目标蛋白上,就像是打上“清除”的标签。这些被打上标签的目标蛋白,也会被送到蛋白酶体复合体处进行降解。
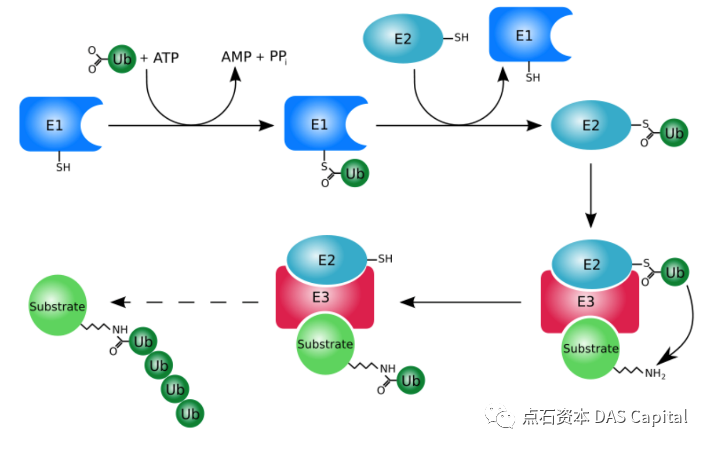
图:泛素化示意图
PROTAC(PROteolysis TArgeting Chimeras,蛋白降解靶向嵌合体)由三种元素组成:E3泛素连接酶配体、靶蛋白配体和Linker。E3泛素连接酶配体负责特异性招募E3泛素连接酶;靶蛋白配体用于靶向和捕获目标蛋白;Linker用于结合这两个配体,形成稳定的三元复合物。
PROTAC分子能够将E3泛素连接酶募集到靶点蛋白附近,为靶点蛋白打上泛素“标签”。在细胞中,打上泛素“标签”的蛋白将被送入蛋白酶体进行降解。靶蛋白降解后,PROTAC分子又可以被释放出来参与到下一个蛋白的降解过程,因此这种降解作用具有催化效果,较少的药物剂量就可以实现高效的降解。
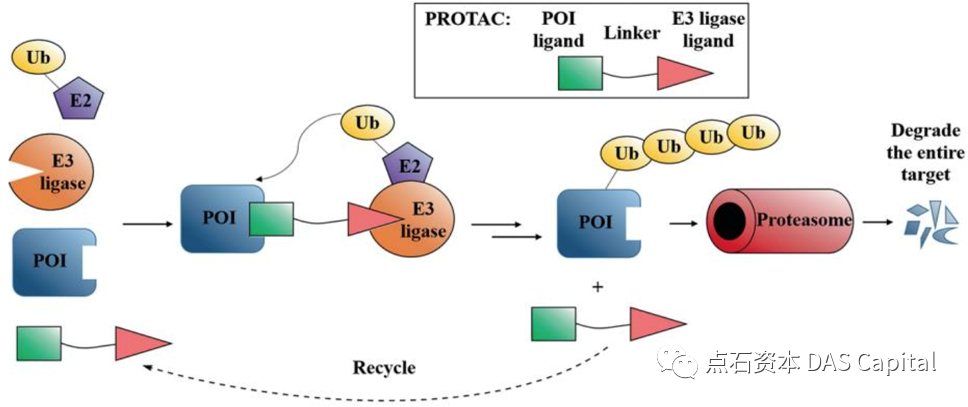
图:PROTAC通过泛素调节的蛋白降解发挥药效
目前,细胞内靶蛋白表达水平的调控主要可以从三个层面进行干预:(1)在基因层面进行靶蛋白的调控技术,例如近年兴起的基因编辑技术(CRISPR-Cas9),优点:精确度高且通用性强等,缺点:不能对靶蛋白进行动态调控,并具有不可逆性以及存在潜在的遗传补偿效应;(2)在转录层面通过RNA干扰技术靶向特定的靶蛋白mRNA,缺点:效率较低,不适用于研究较稳定的蛋白质;
(3)在蛋白层面通过PROTAC 技术进行靶蛋白的化学敲降,该方法具有高效、可逆的优点,并且能够进行催化循环,可以在低剂量下发挥作用
据统计,人类蛋白中有接近80%蛋白无显著活性位点,传统上被认为是不可成药靶点。传统的靶标大部分是具有明确活性位点的蛋白,且适合结合小分子,并主要通过占据活性位点的药理学作用模式 (MOA) 来控制蛋白功能。这种方法虽然简单,但是并不能应用于所有生物靶标,包括那些缺乏酶活、曾被认为是「不可成药」的靶标,以及通过突变反复耐药、现阶段无药可治的靶标。因此长期以来,靶向蛋白的药物研发受限于有限的蛋白种类,极大地限制了药物发展。
自耶鲁大学的Craig Crews教授团队首次报道了小分子PROTAC后的十年间,PROTAC领域飞速发展。药物靶标的格局也因此发生了重大变化,从传统药物靶标转向更具挑战性的靶标。
纵观PROTAC技术发展历史,2001年首个PROTAC分子PROTAC-1诞生,是由PROTAC技术先驱Crews及其同事报道;2008年,第一个小分子PROTAC诞生,同样由Crews课题组设计合成,是由靶向MDM2 E3泛素连接酶的小分子抑制剂nutlin和AR小分子配体以及中间的PEG Linker构成;之后,研究者开发出以CRL4CRBN、CRL2VHL、cIAPE3泛素连接酶配体的各种小分子PROTACs;再之后,Arvinas公司研发的ARV-110和ARV-471两个PROTAC分子进入临床实验,分别用于治疗前列腺癌和乳腺癌。
图:PROTAC里程碑时间点
PROTAC 技术的出现,以其颠覆性的设计、颠覆性的作用机制挑战了「不可成药」靶点,使耐药患者获得新一代治疗药物。近两年来,PROTAC 技术进入高速发展阶段,为生物医药研发开拓了一片新的领域。
蛋白质是细胞内重要的生物大分子,参与多种生命活动,包括酶和激素的功能、运动、运输、免疫反应等。以往,人们非常关注蛋白质在细胞内是如何合成的,但对于蛋白质在细胞内是如何降解的却少有人关注。以色列科学家Aaron Ciechanover、Avram Hershko和美国科学家Irwin Rose发现了泛素介导的蛋白质降解机制,共同获得了2004年诺贝尔化学奖。
泛素本身也是一种多肽,由76个氨基酸残基组成,序列高度保守,且在多种真核生物中普遍存在。泛素的主要功能是标记需要分解的蛋白质,当泛素连接到蛋白上后,会导致这些蛋白被运送到蛋白酶体中进行降解,实现细胞内蛋白的平衡。总的来说,泛素参与了细胞周期、增殖、凋亡、分化、转移、基因表达、转录调节、信号传递、损伤修复、炎症免疫等几乎一切生命活动的调控。
泛素化,主要由3种酶催化,即E1泛素活化酶、E2泛素结合酶、E3连接酶。在ATP的作用下,泛素会被激活,先形成泛素-腺苷酸复合物,再被转移到泛素活化酶E1上;随后,E1将活化的泛素转移到泛素结合酶E2上;最终,泛素连接酶E3将泛素转移到目标蛋白上,就像是打上“清除”的标签。这些被打上标签的目标蛋白,也会被送到蛋白酶体复合体处进行降解。
图:泛素化示意图
PROTAC(PROteolysis TArgeting Chimeras,蛋白降解靶向嵌合体)由三种元素组成:E3泛素连接酶配体、靶蛋白配体和Linker。E3泛素连接酶配体负责特异性招募E3泛素连接酶;靶蛋白配体用于靶向和捕获目标蛋白;Linker用于结合这两个配体,形成稳定的三元复合物。
PROTAC分子能够将E3泛素连接酶募集到靶点蛋白附近,为靶点蛋白打上泛素“标签”。在细胞中,打上泛素“标签”的蛋白将被送入蛋白酶体进行降解。靶蛋白降解后,PROTAC分子又可以被释放出来参与到下一个蛋白的降解过程,因此这种降解作用具有催化效果,较少的药物剂量就可以实现高效的降解。
图:PROTAC通过泛素调节的蛋白降解发挥药效
目前,细胞内靶蛋白表达水平的调控主要可以从三个层面进行干预:(1)在基因层面进行靶蛋白的调控技术,例如近年兴起的基因编辑技术(CRISPR-Cas9),优点:精确度高且通用性强等,缺点:不能对靶蛋白进行动态调控,并具有不可逆性以及存在潜在的遗传补偿效应;(2)在转录层面通过RNA干扰技术靶向特定的靶蛋白mRNA,缺点:效率较低,不适用于研究较稳定的蛋白质;
(3)在蛋白层面通过PROTAC 技术进行靶蛋白的化学敲降,该方法具有高效、可逆的优点,并且能够进行催化循环,可以在低剂量下发挥作用
与传统小分子拮抗剂相比,PROTAC具有一系列独特的优势:
(1)选择性更高、作用靶点更广
PROTAC最大的优势之一是能够使靶点从“不可成药性”变成“可成药性”。多数小分子药物或单抗需要结合酶或受体的活性位点来发挥作用,但据估计,人类细胞中80%的蛋白缺乏这样的位点。PROTAC无需与目标蛋白长时间和高强度的结合,便可捕获蛋白并将其降解,因此有望突破传统难以成药的靶点并克服耐药性问题。
(2)催化降解功能、更低的药物剂量
传统的小分子采用的药理学作用模式为占据驱动模式,为了提高靶点占有率,往往需要高剂量的药物,可能会带来较大的毒副作用。PROTAC采用的是事件驱动(event-driven)的药理学作用模式,其对目的蛋白的降解过程是一种催化作用,因此只需较低的化合物浓度便可以达到很好的降解效率,且能够降解整个蛋白。
(3)延长作用时间
靶蛋白的降解是时间依赖性的,PROTAC可以在几分钟内将胞内靶蛋白消耗到接近基础水平。当原先存在的靶蛋白耗尽后,PROTAC只需降解重新合成的靶蛋白,大多数蛋白质的再合成速度很慢,即使在PROTAC完全清除后,细胞可能仍需要一段相当长的时间,才能将蛋白质恢复正常生理水平,从而大大延长作用时间。

表:PROTAC与其他药物对比
但PROTAC开发难度较大,目前在研的PROTAC存在口服吸收及透膜性较差等问题。一方面,PROTAC药物属于双靶点药物,分子量较大,结构复杂,药代动力学并不乐观,口服吸收和透膜性存在挑战;另一方面,PROTAC靶标蛋白降解能力显著,但由于靶蛋白在正常细胞中也会表达,PROTAC对正常组织的毒性须密切关注。此外,用于小分子药物早期筛选的Lipinski的“五法则”并不适用于PROTAC领域。
目前PROTAC靶点主要分为如RAS、SHP2等难成药类的靶点,以及BTK、EGFR靶向治疗后复发耐药的靶点。
a) 难成药靶点
i. RAS
RAS是癌症中最常见的突变基因,在肺癌、结直肠癌和胰腺癌中均存在过表达或活化。在被发现和研究的40多年来,一直没有针对该靶点的药物上市,长期被称为“不可成药”的靶点。虽然去年5月份上市的AMG510(Sotorasib)终结了该靶点的“不可成药性”,但是降解剂在该靶点仍然大有可为。
针对KRAS-G12C突变,2020年PROTACs技术先驱Craig Crews教授带领的研究小组首次报道了first-in-class的内源性KRAS-G12C降解剂LC-2的开发。研究发现,LC-2可快速、持续地降解纯合和杂合突变细胞系中的KRAS-G12C,导致MAPK信号传导受到抑制,表明PROTACs介导的降解是衰减癌细胞中致癌KRAS水平和下游信号的一种可行策略。
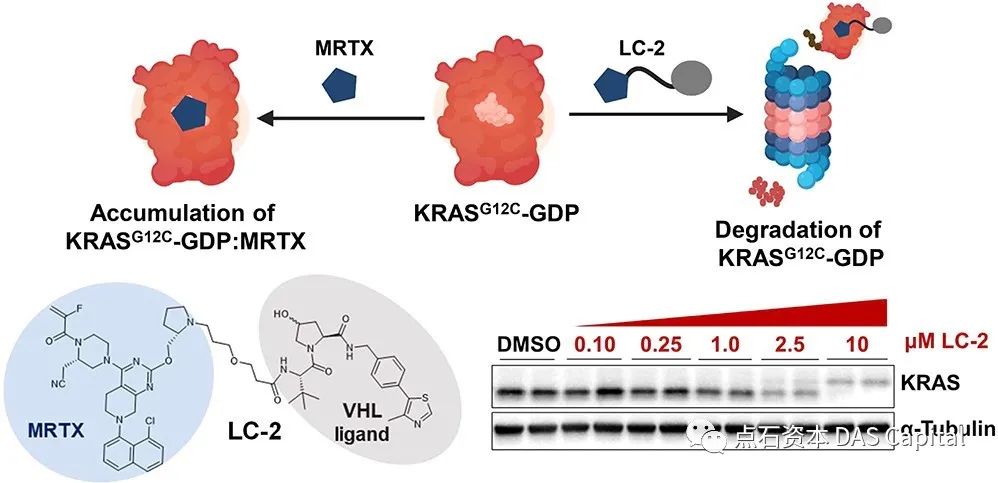
图:首个成功介导内源KRAS G12C降解的PROTAC分子LC-2
Source: ACSCent. Sci. 2020, 6, 8, 1367-1375
可以预见,利用PROTAC技术攻克的将不只是G12C,对G12D等其他突变也将大有可为。
b) 药物导致的突变耐药
小分子抑制剂或拮抗剂在临床用药过程中,不可避免的都会发生获得性耐药。比如EGFR-T790M和C797S耐药等。虽然可以通过开发新一代的抑制剂解决耐药问题,但是随着新一代药物的使用,新的耐药也会随之出现。
i. EGFR
EGFR(Epidermal Growth Factor Receptor)是上皮生长因子(EGF)细胞增殖和信号传导的受体,刺激下游的PI3K/AKT通路、RAS/RAF通路,对细胞的生长、增殖、分化产生重要作用。
EGFR的过度表达和突变与非小细胞肺癌(NSCLC)的发生发展密切相关,也是NSCLC重要的药物靶点之一。目前国内已上市三代EGFR靶向抑制剂,包括(1)第一代EGFR抑制剂:靶向19-Del/21-L858R突变的厄洛替尼、吉非替尼、埃克替尼,(2)第二代EGFR抑制剂:与底物不可逆结合的阿法替尼、达克替尼,(3)第三代EGFR抑制剂:在靶向19-Del/21-L858R突变的基础上同时靶向20-T790M突变的奥希替尼和阿美替尼。但15%~32%的患者会发生第三次EGFR突变,并最常发生在EGFR C797S位点,即奥希替尼与EGFR的结合位点。
面对C797S位点突变,目前临床上尚处于无药可治状态。而利用PROTAC技术开发的EGFR靶向蛋白降解分子有望带来巨大临床获益。
目前,我们每年新发非小细胞肺癌患者人数超过60万人,其中携带EGFR突变的患者占比高达40%。根据弗若斯特沙利文数据,2019年中国EGFR-TKI 药物市场已经接近80亿元人民币,预计2022年中国EGFR-TKI 药物市场规模将超过200亿,CAGR保持30%以上。
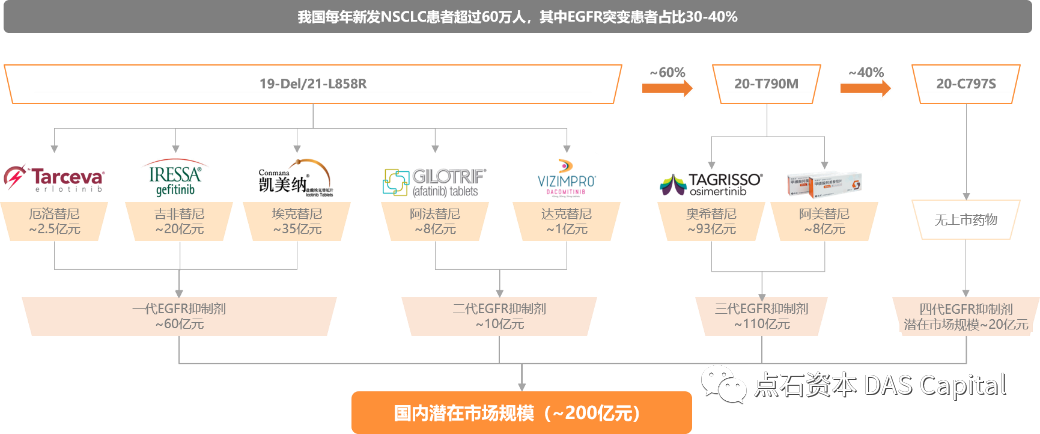
表:非小细胞肺癌EGFR市场规模测算
数据来源:点石整理
Craig M. Crews、金健、张三奇、丁克等课题组都有相关EGFR-PROTACs的研究,研究人员将第一、二、三代抑制剂作为结合靶蛋白EGFR的配体应用到PROTAC的设计中,旨在通过蛋白降解途径克服耐药突变或找寻突破抑制剂的蛋白降解疗法。PROTAC疗法的应用,有望解决用药后EGFR产生次级突变的问题,减少耐药发生,同时能够高效靶向多种EGFR突变,替代一、二、三代EGFR传统小分子抑制剂,推行至一、二线治疗方案。
针对EGFR多突变PROTAC药物的研发并不容易,由于位点选择有限、对选择性与安全性的取舍、合成工艺复杂等多方面因素制约,目前全球尚无产品进入临床阶段。C4 Therapeutics正在开发的候选药物CFT8919靶向EGFR L858R突变的变构位点,临床前数据显示其能显著减少EGFR次级突变,从而减少耐药发生,该药物对C797S和T790M突变同样具有活性,并具有入脑能力,有望对脑转移肿瘤有效。公司计划2022年中申报IND,2022年底开启临床试验。
ii. BTK
BTK激酶是BCR信号通路的关键蛋白,对B细胞的增殖、存活具有重要作用。BTK蛋白缺失会导致成熟B细胞无法产生。伊布替尼作为首个上市的BTK抑制剂,于2013年在美国上市后,一年内便成为销售额增长最快的抗肿瘤药物之一。但是,Ibrutinib也存在两个明显的弱点:一方面是off-target导致的副作用;另一方面是BTK的突变体C481S导致的耐药。
BTK从一代抑制剂到现在的二代抑制剂,虽然药效都非常明显,但极易产生耐药。以一代BTK抑制剂为例,每24小时便有病人的BTK蛋白水平恢复到治疗前的3%至30%以上。
因此,临床急需能够克服BTK突变耐药且具有更小副作用的药物。采用PROTAC技术,一是因为它理论上可以循环利用,当PROTAC进入细胞后,可以持续地降解新生或原有的BTK蛋白;第二,只要依赖BTK,无论是怎样的结合方式,降解之后,其他的作用机制都无法起作用。
(一) 海外

Arvinas由Crews在2013年创立,是最早布局PROTAC的公司之一,开发的蛋白降解技术主要用于肿瘤和神经系统类疾病的治疗。目前,公司已有两款PROTAC药物进入临床II期,分别是靶向雄激素受体(AR)的ARV-110和靶向雌激素受体(ER)的ARV-471。
2021年7月辉瑞与Arvinas达成协议,共同开发并商业化ARV-471。根据协议,Arvinas将获得6.5亿美元的预付款,以及多达14亿美元的里程碑付款,辉瑞还将对Arvinas进行3.5亿美元的股权投资。除辉瑞外,Arvinas还与默沙东、基因泰克、拜耳等制药巨头建立了合作关系。
C4 Therapeutics由James Bradner成立于2015年,该公司拥有专注于蛋白降解剂开发的平台C4T TORPEDO,用于PROTAC的设计、合成和活性评价,旨在发现高质量的蛋白降解剂。
目前布局的靶点与肿瘤相关,如IKZF1/3、BRD9、EGFR、BRAF-V600E和RET等。2019年1月,C4 Therapeutics与渤健和罗氏分别达成4.15亿美元和9亿美元的合作协议。
Kymera Therapeutics成立于2016年,专注于用蛋白降解技术治疗癌症和免疫性炎症,建立了独特的Pegasus平台,该平台可利用生物信息学驱动靶标的发现、建立的E3连接酶库、拥有先进的蛋白降解检测手段和高水准的结构生物学模型。布局的靶点有IRAK4、STAT3等,进展最快的是KT-474,目前在I期临床。
该公司的合作伙伴主要是赛诺菲和葛兰素史克等。2020年7月,与赛诺菲达成多项计划的战略合作,获得1.5亿美元的预付款,并可能获得超过20亿美元的潜在开发、监管和销售里程碑,以及可观的特许权使用费。
(二) 国内
开拓药业(9939.HK)成立于2009年,以雄激素受体(AR)和肿瘤相关疾病为核心,研发多通道产品组合,产品覆盖全球高发病率癌症、新冠、脱发和痤疮等。2021年2月,开拓药业宣布其自主研发的全球首个基于PROTAC技术的外用AR降解剂(GT20029)的IND申请已获国家药品监督管理局受理。该公司将通过开发非口服的PROTAC药物来验证这一开创性的技术是否可以成药,同时也正在研发口服的靶向AR的PROTAC,适应证为雄激素性脱发和痤疮等。
海思科(002653)成立于2000年,公司具有比较完善的PROTAC平台,用以开发针对肿瘤和自身免疫性疾病的高选择性且口服有效的蛋白降解药物。目前PROTAC的项目有超过20个,布局的靶点有BTK、IRAK4、BRD4、AR、EGFR、KRAS、ALK、CDK4/6、EZH2、PARP、Bcr/Abl、STAT3等。
2017年,和径医药在上海科技大学内成立,公司诞生之初就获得了来自联和资本、药明康德、再鼎医药、千骥资本的投资,2018年又宣布携手上海科技大学合作开发“配体导向蛋白质降解药物”,并达成了逾一亿美金的战略合作协议。公司通过与上海科技大学的项目合作起步,在扩大产品管线的同时正在努力打造自己独特的新药研发平台。
和径医药的PROTAC平台起步较早,第一个PROTAC合作项目已经研发超过3年时间,目前已筛选出了具有高成药性的分子系列,进展较快的分子即将开展IND-Enabling。该分子系列具有高口服生物利用度,并在动物体内药效显著,在克服非小细胞肺癌小分子药物耐药等领域处于领先地位。
珃诺生物成立于2018年,是一家专注于小分子创新药研发的生物医药企业。公司专注于开发分子伴侣介导的蛋白质降解(Chaperone-mediated Protein Degradation/Degrader, CHAMPTM)技术平台,目前已有多个全球创新的新一类小分子抗肿瘤化合物即将进入临床或处于临床前开发阶段。
作为革命性技术,PROTAC的发展经历了20年,尤其在过去的5年发展迅猛,俨然已经成为新药研发的新风口。利用该技术可布局的靶点广阔,市场巨大,未来可期。但是PROTAC分子量大、生物利用度差、成药困难的痛点也很明显,期待该技术的不断进步和完善。相信随着成药性差的难题被攻克,PROTAC可以成为像小分子抑制剂、单抗和免疫治疗等一样成功的疗法。当然,也期待看见更多PROTAC药物上市,延长患者生命并提高患者的生存质量。
免责声明
相关内容基于已公开的资料或信息撰写,但本公司不保证该等信息及资料的完整性、准确性,所含信息及资料保持在最新状态。同时,本公司有权对本报告所含信息在不发出通知的情形下做出修改,阅读者应当自行关注相应的更新或修改。
在任何情况下,本篇文章中的信息或所表述的意见均不构成对任何人的投资建议,无论是否已经明示或暗示,本报告不能作为道义的、责任的和法律的依据或者凭证。在任何情况下,本公司亦不对任何人因使用本文章中的任何内容所引致的任何损失负任何责任。本文章仅为本公司所有,未经事先书面许可,任何机构和个人不得以任何形式翻版、复制、发表、转发或引用本报告的任何部分。



{kind=link}